Research interests
For information on Bugbank, a project that aims to integrate infection data into UK Biobank, visit this page.
The goal of my group's research in pathogen population genomics is to reveal the evolutionary forces and phenotypic constraints shaping pathogen genomes by developing innovative new statistical methods based on evolutionary theory, population genetics and computational inference. Our ambition is to contribute towards a complete understanding - evolutionary, epidemiological and molecular - as to why bacteria cause disease so that scientists can reduce the global burden of disease by developing more effective strategies for prevention and treatment. For an introduction to recent advances in the rapidly emerging field of bacterial pathogen population genomics, see for example Wilson (2012) PLoS Pathogens 8: e1002874.
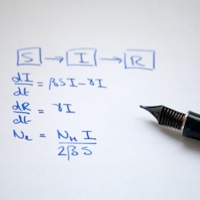
|
Pathogen evolution
The group recently led a study into the evolutionary changes in the major healthcare-associated pathogen Staphylococcus aureus that accompanied the transition from long-term asymptomatic nasal carriage to an acute invasive bloodstream infection within an individual patient. With a whole genome sequencing approach, we discovered that a significant excess of nonsense mutations occurred in the bacterial population colonizing the patient, suggesting that loss-of-function mutations - known to cause potentially radical changes in bacteria - may be associated with pathogenesis within long-term carriers. For more details see Young et al. (2012) PNAS 109: 4550.
Detecting selection
Unearthing evidence of adaptation within the genome can reveal the forces that drive evolutionary change. With Molly Przeworski in Chicago and Gil McVean in Oxford, I have created novel methods - gammaMap and omegaMap - for detecting natural selection in the coding genome both within and between species. These methods have had an impact far beyond just bacteria, revealing insights into the nature of adaptive change in animals, plants and protozoans. The software implementing these methods can be downloaded through this website. For full details of the methods and example applications, see Wilson, Hernandez, Andolfatto and Przeworski (2011) PLoS Genetics 7: e1002395 and Wilson and McVean (2006) Genetics 172: 1411.
Transmission modelling
Transmission history is recorded in the genetic diversity of pathogen populations and may be unlocked through genomic analysis. With Paul Fearnhead at Lancaster, I developed iSource, a method for tracing bacterial transmission from among several potential source populations. We estimated that in England, 97% of cases of campylobacteriosis - one of the leading economically important causes of gastroenteritis - was transmitted from contaminated poultry and meat. Collaborators garnered comparable results in iSource analyses of campylobacteriosis in Scotland and New Zealand. In the latter, this research contributed to a body of evidence supporting the introduction of new controls to the poultry industry that eventually led to a 60% reduction in human disease. The software, described in Wilson et al. (2008) PLoS Genetics 4: e1000203, can be downloaded from this website.
Methods development
My group develops and applies computational and statistical methods to implement novel analysis approaches for pathogen population genomics. Development is a time-intensive process, requiring integration of specifically tailored theory with sophisticated inference. Conceiving, implementing and testing new methods can take months, but we recoup this investment through the release of publically available software and its subsequent use by new collaborators in diverse applications. The group's work has been recognized via citations in F1000 and invitations to group members to talk in numerous countries. For a recent example of our approach to developing new theoretical models that we implement as practical analysis software, see Dearlove and Wilson (2013) Philosophical Transactions B 368: 20120314.



